RODEO
RODEO (Rapid ORF Description & Evaluation Online) is an algorithm to help biosynthetic gene cluster (BGC) analysis, with an emphasis on ribosomal natural product (RiPP) discovery.
RODEO (Rapid ORF Description & Evaluation Online) is an algorithm to help biosynthetic gene cluster (BGC) analysis, with an emphasis on ribosomal natural product (RiPP) discovery.
RODEO (Rapid ORF Description and Evaluation Online) is a tool for evaluation of the local genomic context of protein sequences. RODEO takes as input a list of accession numbers (from GenBank), downloads sequences within a specified distance in the local genomic region, and analyzes the protein sequences for homology to characterized families. Additionally, small peptides that may have eluded annotation in GenBank are translated in all possible frames. The idea behind RODEO is that by rapidly assessing gene neighborhood information, biosynthetic genes can be identified, and predicted properties can be used to guide ribosomal natural product (RiPP) isolation and structure prediction.
RODEO takes advantage of the massive amount of sequence data in the publicly accessible, NCBI-maintained database GenBank (Agarwala et al., 2016). For homology analysis, RODEO uses the HMMER suite (Finn et al., 2015; Finn et al., 2011) to analyze sequences for the presence of profile hidden Markov models (pHMMs) as defined in the Pfam database (Finn et al., 2016). For additional flexibility in analysis, supplemental user-specified pHMMs can also be used to complement the Pfam database.
RODEO's web tool uses MySQL, TORQUE/PBS, and Django and is hosted at the University of Illinois School of Chemical Sciences.
RODEO’s default output consists of an HTML file containing gene tables as well as SVG-format ORF diagrams. Additionally, CSV files of peptides and/or genes can be output for use with analysis of large numbers of biosynthetic gene clusters (BGCs).
The new version of RODEO supports analysis for lasso peptides, class I lanthipeptides, sactipeptides, and thiopeptides.
For certain RiPP classes, RODEO includes an additional likelihood prediction and scoring functionality for the precursor peptides, including leader/core region, molecular weight, etc. We are currently working on generalizing this functionality to even more classes of RiPPs (Arnison et al., 2013).
RODEO can be run natively on a Unix or Mac system or with Unix emulation in a Windows environment, such as Cygwin. In addition to an internet connection, the following must be installed:
See below, "Running on Windows", for some packages that must be installed before the above if you're using Windows/Cygwin.
Most Linux or Mac computers should already have Python installed. See https://www.python.org/ to install it if not. Check for Python (and which version) on the command line via:
python --version
HMMER3 is available from http://hmmer.org/ and on some Linux distributions (e.g. Ubuntu) can even be installed simply by:
sudo apt-get install hmmer
In absence of a package manager (i.e. with certain distributions of Linux or Cygwin), HMM tools can be installed manually by downloading the appropriate binaries from the above URL. These are installed by first unpacking, then making the installer using the following commands.
tar –xf hmmer-3.1b2-cygwin64.tar.gz ./configure make make install
To test if HMMER3 is properly installed, you can type the following to see if the man page appears:
man hmmscan
Biopython can easily be installed using pip
pip install Biopython
Additionally, a copy of the Pfam pHMM database must be downloaded and placed in the rodeo hmm_dir folder (see Installation below). The Pfam database is the only aspect of RODEO of substantial file size, taking up ca. 2 GB of disk space. The pHMMs can be installed using the following FTP commands:
wget ftp://ftp.ebi.ac.uk/pub/databases/Pfam/current_release/Pfam-A.hmm.gz gunzip Pfam-A.hmm.gz hmmpress Pfam-A.hmm
Alternatively, this can be installed manually at http://pfam.xfam.org/, navigating to FTP/current release and downloading the Pfam-A.hmm.gz file, which can be unpacked and pressed in the default RODEO directory with the following command:
hmmpress Pfam-A.hmm
Follow the instructions at the Meme website for a thorough installation guide.
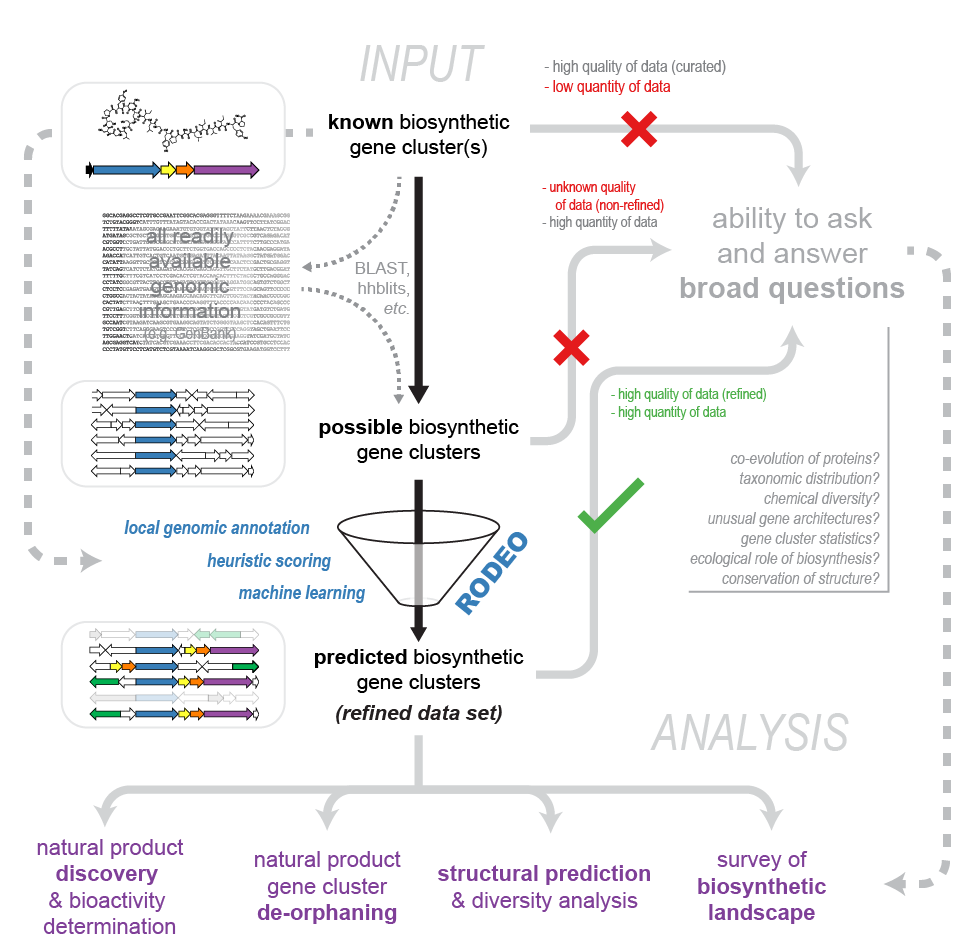
When installing RODEO with Cygwin, ensure that the following packages are installed before installing HMMER, Biopython, etc.:
These can be included during the first installation of Cygwin, or added later by selecting the following packages in the setup executable. At the install prompt, select “Install from Internet”, keep the default directories, and select an installation mirror (we recommend using an academic mirror) and changing the status either to “Install”, “Keep” or the most recent version number (see below).
The archive file can be extracted anywhere. RODEO is run from the extracted directory.
By default, RODEO includes four directories: confs (configuration files), hmm_dir (HMM files), ripp_modules (packages for RiPP class analysis), test_suites (test datasets), and tmp_files (temporary work files).
Installation and use of RODEO on a personal computer requires use of Unix command line. Alternatively, the web tool can be run with only a modern internet browser.
RODEO is run as a Python script within the Bash shell (other shells might work; we haven’t tested these); parameters are input as flags/options in the command line as well as with an optional configuration file. RODEO use takes the general form:
python rodeo_main.py query <options>
RODEO utilizes the following flags:
user@ubuntu:~/rodeo2$ python rodeo_main.py -h
usage: Main RODEO app. [-h] [-out OUTPUT_DIR] [-c [CONF_FILE [CONF_FILE ...]]]
[-hmm [CUSTOM_HMM [CUSTOM_HMM ...]]] [-j NUM_CORES]
[-max PRECURSOR_MAX] [-min PRECURSOR_MIN] [-o OVERLAP]
[-ft FETCH_TYPE] [-fn FETCH_N] [-fd FETCH_DISTANCE]
[-pt [PEPTIDE_TYPES [PEPTIDE_TYPES ...]]] [-ea] [-ex]
[-print]
query
positional arguments:
query Accession number, genbank file or .txt file with an
accession or .gbk query on each line
optional arguments:
-h, --help show this help message and exit
-out OUTPUT_DIR, --output_dir OUTPUT_DIR
Name of output folder
-c [CONF_FILE [CONF_FILE ...]], --conf_file [CONF_FILE [CONF_FILE ...]]
Maximum size of potential ORF
-hmm [CUSTOM_HMM [CUSTOM_HMM ...]], --custom_hmm [CUSTOM_HMM [CUSTOM_HMM ...]]
Maximum size of potential ORF
-j NUM_CORES, --num_cores NUM_CORES
Number of cores to use.
-max PRECURSOR_MAX, --precursor_max PRECURSOR_MAX
Maximum size of potential ORF
-min PRECURSOR_MIN, --precursor_min PRECURSOR_MIN
Minimum size of potential ORF
-o OVERLAP, --overlap OVERLAP
Maximum overlap of search with existing CDSs
-ft FETCH_TYPE, --fetch_type FETCH_TYPE
Type of window specification. 'cds' will make the
window +/- n CDSs from the query. 'nucs' will make the
window +/- n nucleotides from the query
-fn FETCH_N, --fetch_n FETCH_N
The 'n' variable for the -ft=orfs
-fd FETCH_DISTANCE, --fetch_distance FETCH_DISTANCE
Number of nucleotides to fetch outside of window
-pt [PEPTIDE_TYPES [PEPTIDE_TYPES ...]], --peptide_types [PEPTIDE_TYPES [PEPTIDE_TYPES ...]]
Type(s) of peptides to score.
-ea, --evaluate_all Evaluate all duplicates if accession id corresponds to
duplicate entries
-ex, --exhaustive Score RiPPs even if they don't have a valid split site
-print, --print_precursors
Print precursors in HTML file
The -pt flag is for declaring what RiPP classes you'd like to look for. It currently supports lasso peptides (“lasso”), class I lanthipeptides ("lanthi”), sactipeptides (“sacti”), and thiopeptides (“thio”) as arguments. For example,
python rodeo_main.py mixed_types.txt -out mixed -pt lasso lanthi sacti thiowill analyze all queries in mixed_types.txt for lasso peptide, lanthipeptide, sactipeptide, and thiopeptide traits.
Note that when trying to kill RODEO, any signal other than SIGINT will result in potential zombie or orphan processes. The SIGINT signal can be sent by pressing Ctrl+C.
Nearly all parameters can also be supplied in a configuration file. By default, RODEO uses the configuration file in confs/default.conf . This is simply a text file whose syntax will be shown below
Configuration file syntax:>[Ripp_type or 'general'] #BEGIN_VARIABLES [variable_type] VARNAME1 VALUE1 ... ... [variable_type] VARNAMEn VALUEn #END_VARIABLES #BEGIN_COLORS HMM_ANNOTATION_ID1 [color1] ... ... HMM_ANNOTATION_IDm [colorm] #END_COLORS >>
Note that for colors, the common names from the SVG Specifications (click that link for a list of 147 names) can be used, but any hex code will also be accepted (including the #). For instance, entering either royalblue or #4169E1 will give the same color.
Often one will want to supply their own pHMMs to supplement those within the Pfam database. By default, RODEO will scan proteins for all pHMMs included in whatever files are specified after the -hmm flag (see above); the Pfam database is still scanned, as well.
We have found that HMMER3 is good for generating pHMM files from multiple sequence alignments using hmmbuild. See the HMMER3 documentation for how to do this. Alternatively, existing pHMMs can be downloaded from other databases, such as TIGRFAM (Haft et al., 2013; Haft et al., 2003).
Once you have a desired pHMM file, use the HMMER3 command hmmpress <filename> on your .hmm file in order to convert it to the binary format required for use with hmmscan (and, by extension, RODEO). This will generate in the same folder 4 additional files with the same root name but different file extensions.
hmmpress <filename>
In the event that some aspect of RODEO (or all of it) isn’t working, please contact Prof. Douglas Mitchell.
If things aren’t working, though, first consider the following:
Also, please note that since RODEO depends on fetching sequence data from GenBank, the status of the NCBI servers impacts whether the program will run. Off-peak hours (9 pm to 9 am or so EST) seem to be the least error-prone. If you see a string of ‘no results’ errors, it is most likely the case that the servers are having issues; try again in several hours.
The new release of RODEO was developed by Bryce Kille and was designed by Jonathan Tietz, Christopher Schwalen, and Douglas Mitchell. The original RODEO which was written in Perl was developed by Parth Patel.
RODEO's web tool was designed and coded by Kisurb Choe and hosted at the University of Illinois School of Chemical Sciences.
We are grateful to numerous members of the Mitchell Lab, particularly Graham Hudson, Brandon Burkhart, and Xiao Rui Guo, for helpful feedback.